Nucleation
Crystallization from a supersaturated solution frequently proceeds through a series of transient, metastable phases prior to the formation of the lowest-energy, stable polymorph. This phenomenon, popularly referred to as “Ostwald’s Rule of Stages”, has been observed across nearly all classes of solid-state materials: from inorganic minerals and functional technological materials to organic crystals and biological protein crystals. Despite there being a lower thermodynamic driving force for the formation of a metastable polymorph, a metastable phase can still dominate the kinetics of crystallization if it has a lower nucleation barrier than the equilibrium phase. Following the complete crystallization of a metastable phase, the next more-stable polymorph nucleates and grows – consuming the previous metastable phase in a recursive, energetically-cascading series of polymorphic stages down to the lowest-energy, equilibrium phase.
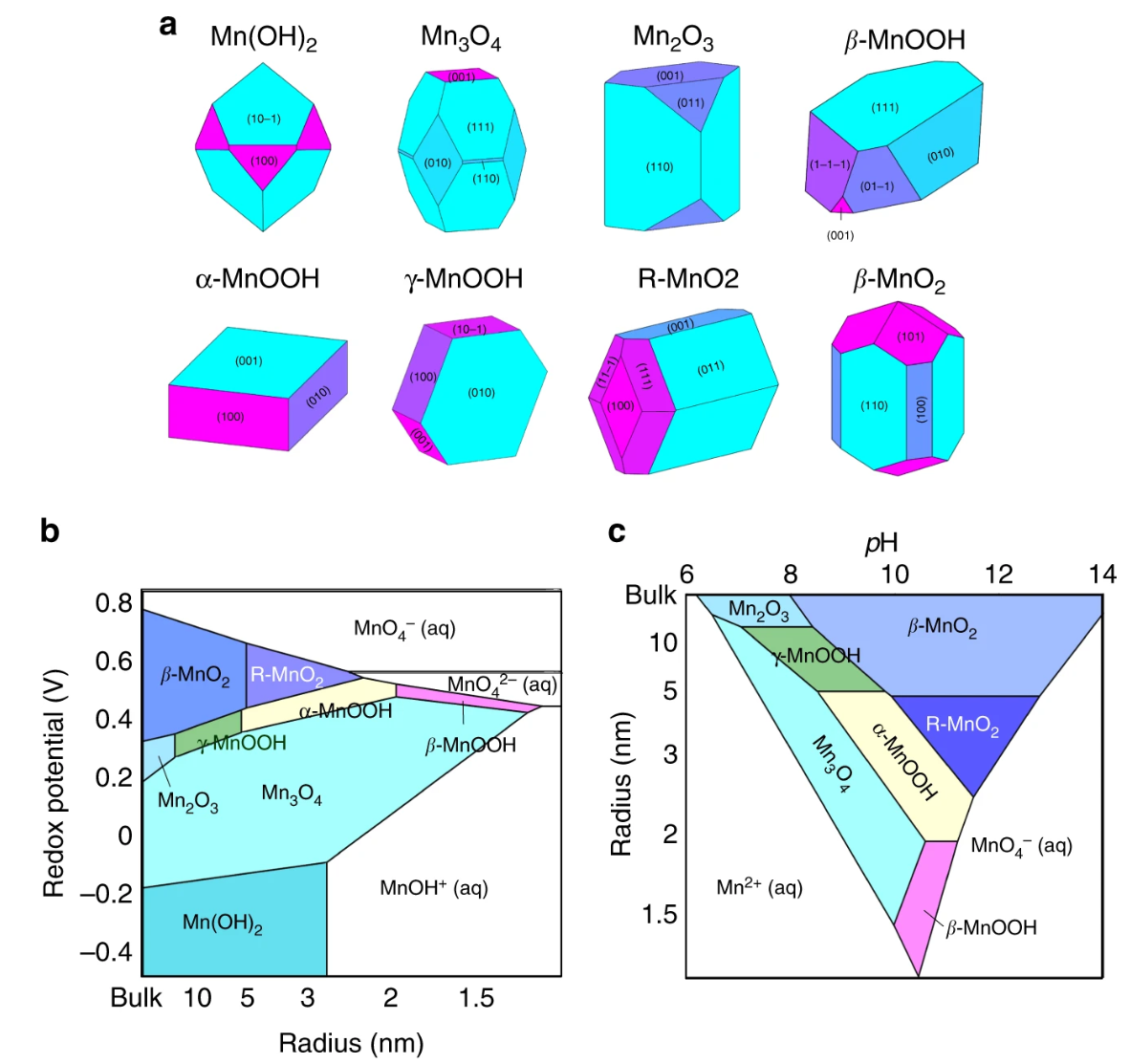 Calculated nanoparticle shapes of various manganese (hydr)oxides. Nanoscale aqueous stability diagrams calculated by combining these surface energies with E-pH (Pourbaix) diagrams
Calculated nanoparticle shapes of various manganese (hydr)oxides. Nanoscale aqueous stability diagrams calculated by combining these surface energies with E-pH (Pourbaix) diagrams
By computing the energy of a critical nucleus in equilibrium with an aqueous environment, we can model how pH, redox potential, and alkali ions can influence the relative nucleation rates between competing polymorphs. We developed new high-throughput tools to compute the surface energies of various crystal structures using DFT. By combining these surface energies with bulk free energy calculations solid-aqueous equilibria, we built a theoretical framework that can explain why the high-pressure Aragonite polymorph of CaCO3 nucleates under ambient conditions in seawater, and can rationalize the multistage crystallization of manganese oxide polymorphs in solution. Our computed ‘synthesis maps’ successfully capture which metastable polymorphs nucleate first, the order of their appearance, and their relative lifetimes.
- Efficient Creation and Convergence of Surface Slabs, Surface Science (2013)
- Nucleation of metastable aragonite CaCO3 from seawater, PNAS (2015)
- Induction Time of a Polymorphic Transformation, Crystengcomm (2017)
- Non-equilibrium crystallization pathways of manganese oxides in aqueous solution, Nature Communications (2019)